
Background
In 2020, the European Medicines Agency (EMA) published EMA Regulatory Science to 2025, which included among its top five core recommendations the promotion of the use of high-quality real-world data in regulatory decision making [1]. The OPTIMAL framework is a set of criteria developed by the EMA for the regulatory use of RWE, which addresses its operational, technical and methodological challenges [2]. Although there is recognition of the need to include RWE in regulatory submissions, and efforts have been made to define the bounds of its appropriate use, the development of firm guidelines remains a strategic priority [1, 2]. As such, RWE is not yet widely provided alongside clinical trials in regulatory submissions, with its inclusion commonly limited to post-approval research and safety monitoring [3, 4]. However, the recent evidence proposing the medicine adoption model has suggested that the inclusion of RWE at the regulatory stage may increase the depth of maximal adoption by 31 patients for every 100 trial patients and decrease the time to maximal adoption by 22 months for every 100 trial months [5]. This shows the potential for profound benefits resulting from the early generation of RWE alongside clinical trials for both pharma and patients.
Based on the systems theory [6], the medicine adoption model has suggested that the approval, reimbursement, prescription and receipt of medicines occurs in an open system comprising three sequential subsystems involving regulators, payors and prescribers [5]. Each subsystem requires a specific set of evidence upon which to base their decisions. This is normally provided in the form of randomised-controlled trial evidence (RCTE) and stakeholder-specific RWE. This evidence is appraised by each subsystem, and if its internal logic is satisfied, and the output is favourable, then the medicine undergoing approval progresses to the next system. In the last subsystem, the prescriber approves the decision to treat patients with the medicine, at which point, patients receive it. Thus, for medicines to reach patients through this system, the evidence provided to gatekeeper stakeholders must answer the research questions asked by each subsystem.
The three subsystems can be considered heterogeneous in their attitudes to RWE and therefore require a multiple stakeholder approach to RWE generation [7] to ensure the timely progression of a medicine to appropriate patients. Put simply, a reliance on RCTE alone at any stage of adoption is likely to be suboptimal. In addition, this research suggested that the prescriber controls the time to maximal adoption of a drug and that the expected increase in the depth of maximal adoption was due to the propensity of RWE to represent a greater proportion (breadth) of the disease population [7]. Failing to utilise RWE to its greatest benefit at the time of regulatory submission may, therefore, prevent an indicatable population from receiving a drug from which it may benefit. The proportion of the disease population indicated for treatment with a new medicine is defined by the regulator subsystem, which has traditionally been supplied with RCTE alone, despite its well-documented limitations regarding its generalisability to the real world. The additional influence of RWE on this subsystem and its output of indicated proportion of a disease population is the subject of this research.
Methods
Study aims
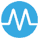
The primary study aim was to determine whether the addition of RWE to RCTE during regulatory engagements for clinical trials increased the breadth of the population indicated to receive rare drug medications as compared to that indicated by regulatory engagements during which RCTE was submitted alone. We expected a directly proportional effect between the sizes of the respective indicated populations. If we were unable to determine this, then we wanted to know when this effect would likely be detectable given the current rate of increase in the addition of RWE to RCTE during regulatory engagements. The secondary aim was to determine whether the indicatable population in RWE submitted during regulatory engagements was broader than that in the RCTE it accompanied to determine the underlying mechanism for any possible effect (Fig. 1).
Fig. 1
The medicine adoption model: PH − (non-pharmaceutical company stakeholder groups); PH + (pharmaceutical company stakeholder groups)

Design, materials and processes
This research was an analysis of data concerning all orphan drugs approved between 2009 and 2019 on the European Medicines Agency (EMA) Orphan Register. Between September and December 2019, a review of the specified engagements was conducted. An engagement comprised an individual submission of evidence supporting a medicine by the marketing authorisation applicant/holder (MAA/H) to the EMA and the EMA’s subsequent response. We categorised the constituent evidence as either RWE or RCTE. Then, we determined the size of indicatable population (the therapeutic indication requested by an engagement) and indicated population (the therapeutic indication ultimately granted) for each type of evidence by measuring the frequency count of criteria limiting the types of patients who could receive a drug. For example, the indicatable population in the first engagement for Adcetris included patients with relapsed or refractory Hodgkin lymphoma and those with relapsed or refractory systemic anaplastic large cell lymphoma (criteria count = 4). It was assumed that more limiting criteria represented a narrower indicatable/indicated population. The indicatable population included limiters extracted from both RCTE and RWE when available (Fig. 2). Engagements were excluded if they contained only RWE, if they contained neither RCTE nor RWE or if information on proposed or indicated populations was unavailable. Each engagement was considered conditionally independent for drugs with more than one engagement due to the supporting evidence related to a separate proposed population.
Fig. 2
Schematic diagram of the analyses conducted to answer the research question
Statistical analyses
A bar plot visualised the distribution of engagements and types of evidence they contained. A chi-square test was conducted to assess the association between the indicated population and the type of evidence (RCTE with or without RWE). A Wilcoxon’s rank sum test assessed the difference in the frequency count of limiting criteria between RCTE and RWE studies. For all analyses, p values ≤ 0.05 were considered statistically significant, and R version 4.0.2 was used. A power analysis evaluated the influence of the small sample size of RWE studies on the overall results. Prediction modelling extrapolated these results to a level expected to deliver significance and the time this would take. The modelling based on its accrual rates of future studies (or engagements) on the increase of studies from 2010–2014 to 2015–2019 and assumed the rise in RCT studies was linear. The model considered the increase in future RWE in both linear and exponential settings. The results presented the power required to detect the observed difference in the indicated population between the RCTE and RCTE + RWE groups as constant.
Results
The review identified 103 engagements in total, of which three were excluded (one contained only RWE; two contained only systematic literature reviews), leaving 100 engagements for 87 different orphan medicines in the final analysis. Of the 87 medicines included, 11 had more than one engagement. Of the 100 engagements included, 87 consisted of only RCTE, 13 consisted of RWE and RCTE, and one consisted of only RWE. Figure 3 provides the distribution of number of engagements for each drug, types of study included in engagements and the overall distribution of study types. Table 1 provides the difference in indicated population for each type of evidence included in engagements (RCTE with or without RWE). The findings showed a broader indicated population in 49 of 87 engagements (56.32%) in which RCTE was submitted alone compared to 10 of 13 engagements (76.92%) in which RCTE was submitted alongside RWE. However, this observed difference was not statistically significant (p = 0.269).